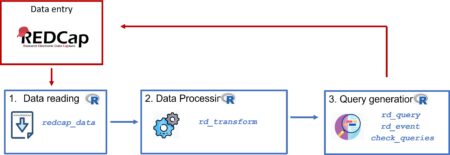
Introduction
If you’re an R user, you’ve probably experienced these moments:
- You’re writing code and forgot the exact syntax for a function
- Your code throws an error and you’re staring at a confusing error message
- You have a block of code but want to understand what it does in plain English
- You want to chat with an AI assistant about your data analysis, but don’t want to leave RStudio
llmcoder is an RStudio addin that solves all of these problems by integrating Large Language Model (LLM) assistance directly into your RStudio workflow, and more importantly, it’s FREE!
In this post, I’ll show you how llmcoder can speed up your R coding and make your workflow smoother.
Watch a quick demo of llmcoder in action:
Installation
You can install llmcoder from GitHub:
# Install remotes if you haven't already
install.packages("remotes")
# Install llmcoder
remotes::install_github("ShiyangZheng/llmcoder")
Load the package:
library(llmcoder)
Feature 1: Generate R Code from Inline Comments
Ever wish you could just type what you want in plain English and get R code instantly?
How to use:
- Type a comment describing what you want
- Place your cursor on that line
- Use the Addins menu and select “Generate Code from Comment”
Example:
# Load the mtcars dataset and create a scatter plot of mpg vs wt, colored by number of cylinders
After running the addin, the comment is replaced with:
library(ggplot2)
data(mtcars)
ggplot(mtcars, aes(x = wt, y = mpg, color = factor(cyl))) +
geom_point(size = 3, alpha = 0.8) +
labs(
title = "Fuel Efficiency vs Weight by Cylinder Count",
x = "Weight (1000 lbs)",
y = "Miles per Gallon",
color = "Cylinders"
) +
theme_minimal()
No more switching to ChatGPT or copying code from Stack Overflow!
Feature 2: Fix Console Errors with LLM Assistance
We’ve all been there – a cryptic error message and you’re not sure what went wrong.
How to use:
- Run code that produces an error
- The error appears in the console
- Use the Addins menu and select “Fix Error with LLM”
Example:
library(dplyr) data %>% filter(cyl == 4) %>% summary() # Error: object 'data' not found
llmcoder captures the error and sends it to the LLM, which returns an explanation and suggests:
mtcars %>% filter(cyl == 4) %>% summary()
Feature 3: Explain Selected Code in Plain English
Sometimes you inherit code from a colleague or find a Stack Overflow answer and want to understand what it does.
How to use:
- Select a block of code in the editor
- Use the Addins menu and select “Explain Code”
Example:
mtcars %>%
group_by(cyl) %>%
summarize(
mean_mpg = mean(mpg, na.rm = TRUE),
sd_mpg = sd(mpg, na.rm = TRUE),
count = n()
) %>%
arrange(desc(mean_mpg))
llmcoder returns:
- Takes the built-in
mtcarsdataset - Groups the data by the number of cylinders (
cyl) - Calculates the mean and standard deviation of miles per gallon (
mpg) for each group - Arranges the results in descending order of mean fuel efficiency
Feature 4: Multi-Turn Chat Panel with Session Context
This is the flagship feature. llmcoder includes a Chat Panel that understands your current R session.
How to open: Use the Addins menu and select “Open Chat Panel”
What makes it special?
The Chat Panel is session-aware:
- It knows which packages you have loaded
- It knows what objects are in your global environment
- It can read the contents of your current script
- It has access to your recent console history
Example conversation:
You: What’s the correlation between mpg and wt in mtcars?
AI: The correlation between mpg and wt in the mtcars dataset is -0.87, indicating a strong negative relationship. As weight increases, fuel efficiency decreases.
cor(mtcars$mpg, mtcars$wt, use = "complete.obs")
Want to see the Chat Panel in action? Watch this demo:
https://youtu.be/zP-RuCN3q14
Supported LLM Providers
llmcoder supports multiple LLM providers – you can choose the one that works best for you:
| Provider | API Key | Notes |
|---|---|---|
| OpenAI (GPT-4/3.5) | Yes | Most popular |
| Anthropic (Claude) | Yes | Great for long conversations |
| DeepSeek | Yes | Cost-effective |
| Groq | Yes | Very fast inference |
| Together AI | Yes | Open-source models |
| OpenRouter | Yes | Access multiple models |
| Ollama | No | Fully local, no API key! |
| Custom endpoint | Yes | LM Studio, vLLM, llama.cpp |
Privacy note: If you use Ollama, all processing happens locally on your machine. No data is sent to external servers.
Customization: Choose Your Prompt Style
The Chat Panel allows you to select different prompt styles:
- General Assistant: Best for general questions
- R Code Helper: Focuses on writing clean, idiomatic R code
- Statistics Advisor: Helps with statistical concepts and test selection
- Research (Psycho): Tailored for psycholinguistics researchers
Why llmcoder?
There are many AI coding assistants out there (Copilot, Cursor, etc.), so why llmcoder?
- Native RStudio integration: No need to switch to another app or browser tab
- Session-aware: The LLM knows what you’re working on
- Multiple LLM providers: Choose the one you prefer (or use a local model for privacy)
- Open source: MIT license, free to use and modify
- Designed for R users: Not a generic coding assistant – it understands R-specific workflows
Call to Action
Ready to try llmcoder?
remotes::install_github("ShiyangZheng/llmcoder")
GitHub: https://github.com/ShiyangZheng/llmcoder
If you encounter any bugs or have feature requests, please file an issue: https://github.com/ShiyangZheng/llmcoder/issues
Star the repo if you find it useful!
About the Author
Shiyang Zheng is a PhD student in Psycholinguistics at the University of Nottingham. His research focuses on idiom acquisition and computational modeling. He built llmcoder to make R coding easier for himself and the R community.
- GitHub: @ShiyangZheng
- Academic website: shiyangzheng.top
- ORCID: 0000-0003-0511-4683